3.2. Transport of glutamine and glutamate across the blood-brain barrier
20-50% of plasma ammonia circulates through brain capillary cells and is incorporated rapidly into the amide group of glutamine by astrocytes. Even at physiological plasma ammonia levels this would raise osmolality and cause cell swelling. However, cell swelling only occurs during hyperammonaemic episodes, i.e. when glutamine is formed at a high rate in astrocytes. Which mechanism protects the brain against astrocyte cell swelling at physiological plasma ammonia concentrations? To answer this question we need to focus on transport processes of glutamine and glutamate. We have already discussed the transport of these amino acids within the glutamate-glutamine cycle, i.e. between neurons and astrocytes. To decrease the cerebral concentrations of these amino acids, glutamine and glutamate need to be transported to the blood across the blood-brain barrier.
To understand the glutamine and glutamate transport across the blood-brain barrier, please study the following figure and read the corresponding paragraphs in ref. 10
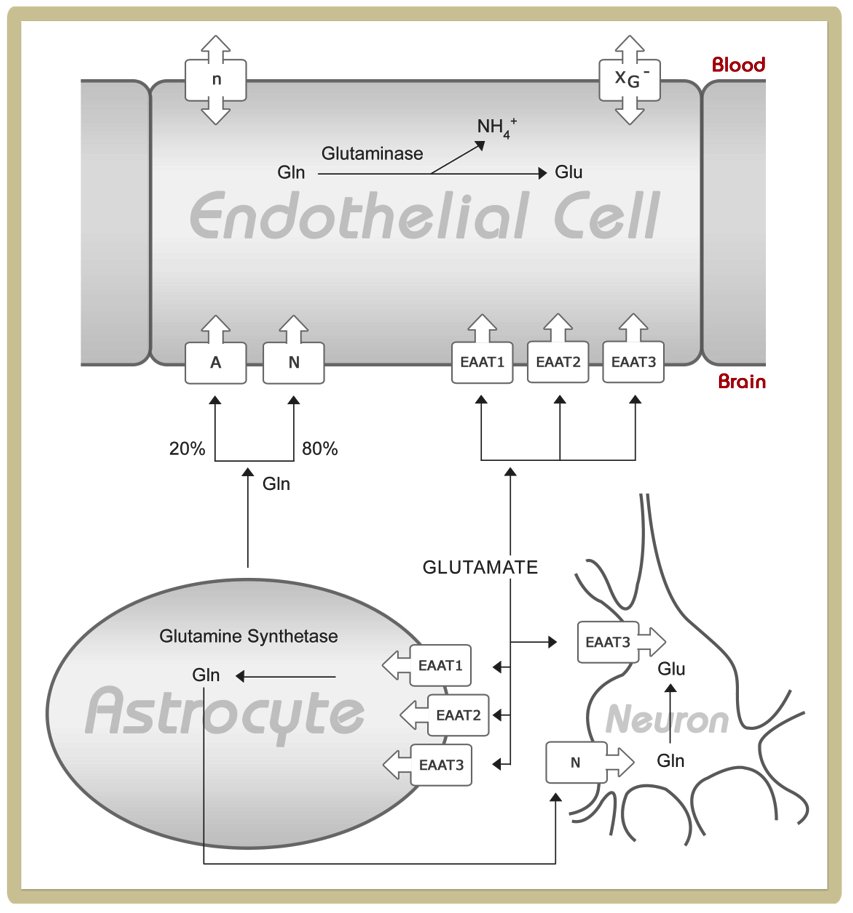
Abbreviations:
A, system A; EAAT, excitatory amino acid transporter; Gln, glutamine; Glu, glutamate; n, system n; N, system N; xG-, system xG-
Source: http://jn.nutrition.org/content/136/1/218S.full
It is important to note that brain capillary endothelial cells, that are rich in tight junctions and thus form the blood - brain barrier, are polarized cells. This means that transporters are not necessarily expressed at the same density on the luminal and abluminal membrane. Furthermore, different types of amino acid transporter may be expressed at the luminal and abluminal membrane:
1) Facilitative amino acid transporters mediate transport along naturally occurring gradients. Since gradients may be varied (e.g. by postprandial increased plasma concentrations) the direction and intensity of the net flux may fluctuate.
2) Sodium-dependent amino acid transporters mediate co-transport of amino acids and sodium along a sodium gradient. The sodium gradient is built up by Na+/K+-ATPases.
Based on the expression pattern and density of transporters (luminal and/or abluminal membrane), their kinetic properties and gradients between plasma and brain tissue and the likelihood of influx or efflux transport at the blood-brain barrier differs significantly among transportable hydrophilic compounds.
The expression pattern of glutamine and glutamate transporters at the blood-brain barrier has functional consequences. The lack of a facilitative carrier for glutamate and glutamine at the abluminal membrane prevents glutamine and glutamate influx to the brain occurring at a high rate if plasma levels of these amino acids increase. Since glutamine and glutamate are actively pumped from the extracellular compartment of the brain to endothelial cells by abluminal sodium-dependent transporters (A, N, EAAT1-3), the intracellular concentrations of these amino acids increases in endothelial cells. If glutamine and glutamate concentrations in endothelial cells raise above those found in plasma, passive transport along this gradient occurs via a facilitative carrier located at the luminal membrane. Taken together, combined transport of glutamine and glutamate across the blood-brain barrier allows removal of access glutamine from the brain and thus prevents astrocyte cell swelling and brain oedema at physiological ammonia and glutamine concentrations.
During hyperammonaemic episodes in patients with urea cycle defects a large proportion of de novo synthesised glutamine is trapped in the brain due to excess formation and limited transport of glutamine across the blood-brain barrier. In addition, disruption of the glutamate-glutamine cycle results in energy impairment and excitotoxicity.
In patients with secondary dysfunction of the urea cycle such as propionic or methylmalonic aciduria glutamine transport across the blood-brain barrier is not inhibited by high plasma glutamine concentrations since these patients develop normoglutaminergic hyperammonaemia. Normal plasma glutamine concentrations in these patients reflect limited availability of 2-oxoglutarate and glutamate due to limited flux through the TCA cycle. It has been shown that propionyl-CoA not only inhibits N-acetylglutamate synthase (causing hyperammonaemia) but also pyruvate dehydrogenase complex (causing lactic acidemia and energy impairment). Furthermore, 2-methylcitrate, which is formed from condensation of propionyl-CoA and oxaloacetate, blocks three enzymes of the TCA cycle. This limits the ability to maintain adequate levels of glutamine precursors. Accordingly, patients with propionic and methylmalonic acidurias may be at a lower risk for brain oedema than patients with urea cycle defects. On the other hand, hyperammonaemia may aggravate pre-existing cerebral energy deficits in patients with propionic and methylmalonic aciduria and thus contribute to the development of “metabolic strokes” in the basal ganglia, though as yet there is little direct evidence for this.
Footnote
Deficiency of another cerebral glutamate transporter, i.e. the mitochondrial aspartate-glutamate carrier isoform 1 (AGC1) has been described. This transporter is specific to neurons and muscle but is not expressed in liver. AGC1 is as a component of the malate-aspartate shuttle providing energy for neurons in the CNS via mitochondrial oxidation of cytosolic NADH. Reference 11 gives details of the subject.
Interestingly, this disease is different from AGC2 deficiency (better known as citrin deficiency or citrullinaemia type 2) which is characterized by hyperammonaemic episodes and a liver-specific impairment of argininosuccinate synthetase secondary to the lack of one of its substrates, aspartate.