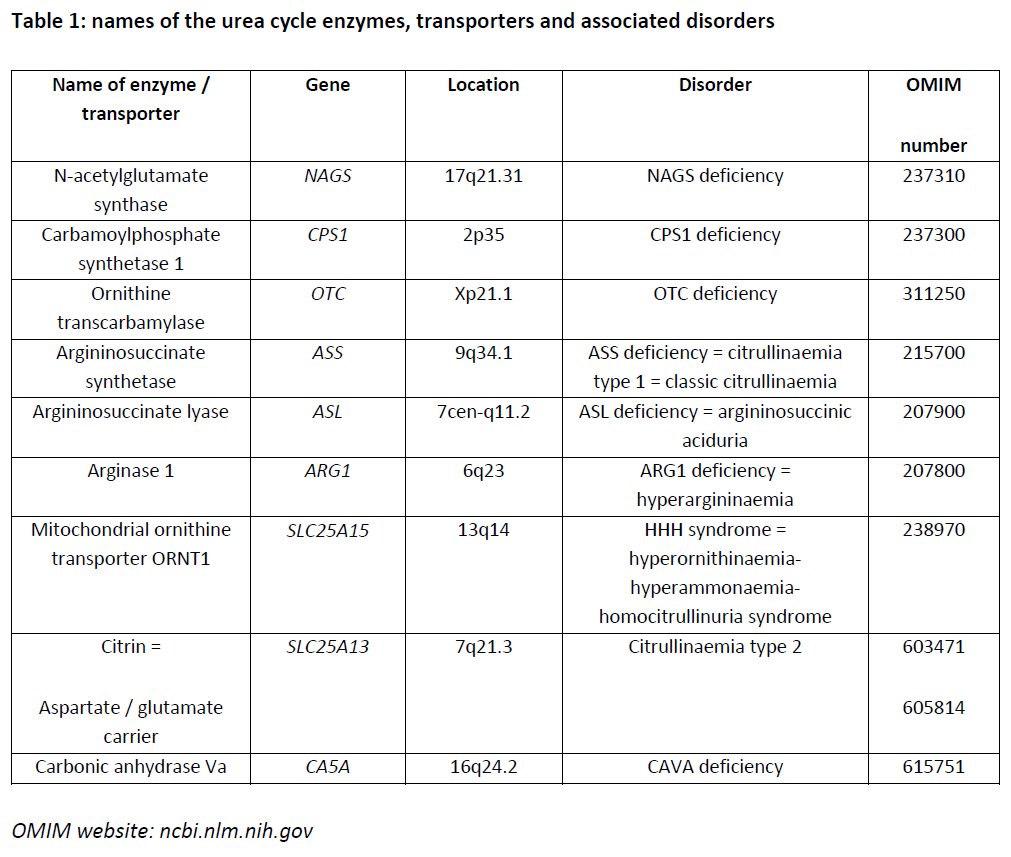
1. Basic Concept - The Urea Cycle
The urea cycle, elucidated by Krebs and Henseleit in 1932, was the first metabolic cycle described in mammals.
The urea cycle is only fully expressed in periportal hepatocytes and consists of two transporters and five enzymes plus a further enzyme (N-acetylglutamate synthase, NAGS) that produces the allosteric activator (N- acetylglutamate, NAG) of the first urea cycle enzyme CPS1 (Figure 3). Three of the enzymes, namely NAGS, CPS1 and OTC are localized in the mitochondrion while ASS, ASL and ARG1 are cytosolic enzymes. Hence, a transporter is required to carry the intermediates ornithine and citrulline in both directions across the mitochondrial membrane (mitochondrial ornithine transporter ORNT1). Yet another transporter is required for a normal urea cycle function—the liver aspartate / glutamate carrier citrin that provides aspartate as a substrate of ASS. The urea cycle also requires carbonic anhydrase Va (CAVA) to form bicarbonate within mitochondria. Defects of all the urea cycle enzymes as well as the transporters exist. Some of these defects cause hyperammonaemia.
The following table lists the names of the urea cycle enzymes and transporters, the name of genes and their chromosomal location, and the associated disorders with OMIM numbers.
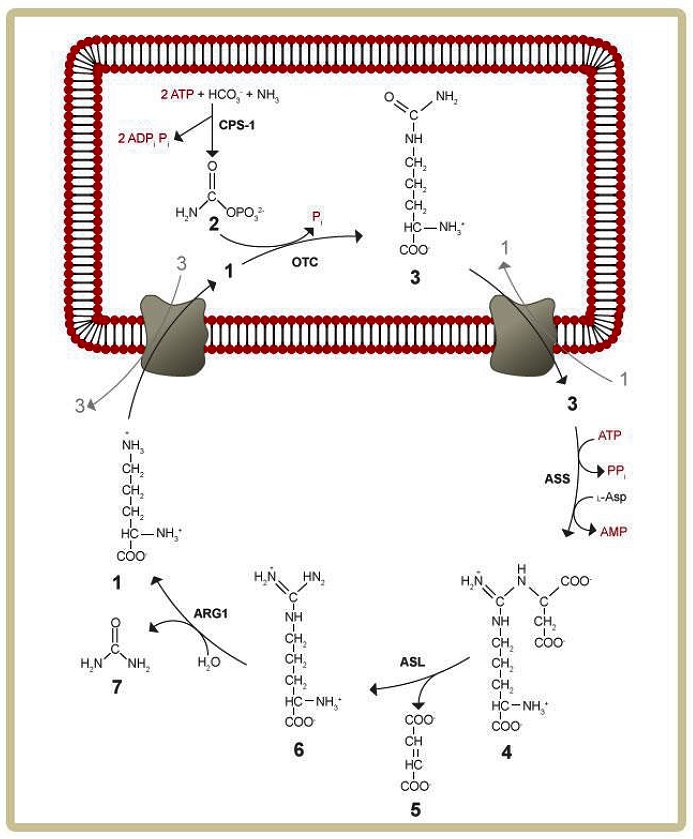
Source: adapted from http://www.answers.com/topic/urea-cycle
Legend: CPS 1: Carbamoyl - phosphate synthetase; OTC: Ornithine transcarbamylase; ASS: Argininosuccinate synthetase; ASL: Argininosuccinate lyase; ARG1: Arginase; 1 Ornithine; 2 Carbamoyl phosphate; 3 Citrulline; 4 Argininosuccinic acid; 5 Succinate; 6 Arginine; 7 Urea.
There are two different liver cell compartments providing different pathways for nitrogen metabolism: the periportal hepatocytes and cells localized in the perivenous compartment.
Ref 2 provides more details and explains why there is division of function between the two cell types as well as giving more valuable information regarding nitrogen and ammonia metabolism. In addition it describes acid/base homeostasis and glutamate cycling.
Nutritional protein is absorbed as polypeptides and amino acids in the intestine and then transported in portal blood from the intestine to the liver with glutamine as the main nitrogenous transport vehicle. Entry into the liver is via the portal vein and thus nitrogenous compounds will first be in contact with periportal hepatocytes. These cells have high expression of urea cycle enzymes, allowing most waste nitrogen to be converted to urea. Before blood returns to the circulation via the hepatic vein, most of the remaining ammonia is incorporated into glutamine by the perivenous hepatocytes, which express a high level of the enzyme glutamine synthetase (see also 1.4 below).
In addition to liver, part of the urea cycle is expressed in the small intestine and kidney. Renal epithelial cells also hydrolyse glutamine to form ammonia that is subsequently excreted in the urine as NH4+. The primary purpose of this so-called renal ammoniagenesis is to allow the excretion of acid for acid-base homeostasis.
The urea cycle serves mainly to detoxify excess nitrogen which cannot be metabolized to other biological substances and would therefore lead to an increase of ammonia above the upper limit of normal (a situation called hyperammonaemia). The urea cycle secondarily produces arginine which would otherwise be an essential amino acid. Finally, urea synthesis serves acid-base homeostasis because large amounts of bicarbonate, produced during protein catabolism, are removed.
Detoxifying ammonia within the urea cycle consumes three molecules of adenosine-triphosphate (ATP), two for the reaction of CPS1 and one for the reaction of ASS. However, the urea cycle releases energy through glutamate dehydrogenase and malate dehydrogenase which reduce NADH, yielding an overall net gain of energy for ATP production.
Fig 3 above indicates the ATP consumption.